Research
Overview
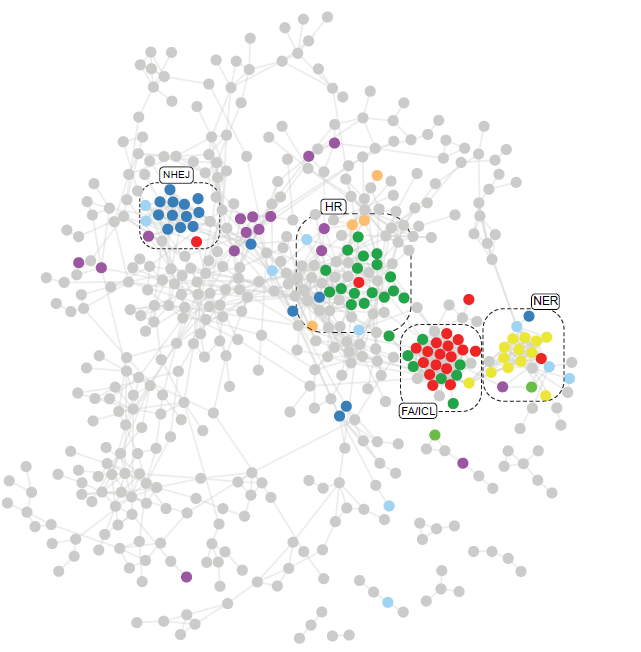
Figure from Olivieri et al. 2020
The major objective of the work done in our laboratory is to understand the processes by which cells maintain the stability of their genome, and to develop therapeutic strategies that exploit this knowledge. We are using genetic, biochemical, functional genomic and proteomic approaches as well as collaborating with structural biologists, computational biologists, and mouse geneticists to achieve these goals. We are also aiming to translate our insights into therapies.
There are three main disease areas where knowledge of genome maintenance mechanisms can inform human health: oncology, immunology and neurobiology. DNA damage is also likely to be a major contributor to age-related tissue dysfunction.
DNA damage and its repair plays a critical role in molecular oncology, as cancer can be viewed as a disease of the genome where mis-repaired DNA damage leads to the mutations and genome alterations that under malignant growth. Multiple DNA damage repair and signaling factors act as tumor suppressors. Prime examples include the BRCA1 and BRCA2 genes, whose mutations increase the risk of breast, ovarian, prostate, and pancreatic cancers. BRCA1 and BRCA2 act in DNA repair by promoting homologous recombination. Furthermore, many classical cancer therapies have the induction of DNA lesions, in particular DNA double-strand breaks, as their mechanism of action. Some targeted cancer therapies exploit DNA repair defects in cancers. Indeed, the inhibitors of the single-strand break repair enzyme, PARP, are highly effective in homologous recombination-defective cancers by exploiting the concept of synthetic lethality (Setton et al, 2021). The critical importance of DNA damage and its repair for cancer initiation, development and therapy motivates us to unlock the mysteries of this fascinating biological process.
Adaptive immunity mediated by B and T cells require programmed genome rearrangements for generating the diversity in T cell receptors and immunoglobulins that are necessary to fight off pathogens and promote anti-tumor immunity. These genome rearrangements, V(D)J recombination and immuglobulin class switching involved the formation of DNA double-strand breaks and their repair. Defective DNA repair or response to DNA damage is often associated with immunodeficiency. Recent work has also linked defective DNA repair with innate immunity. Indeed, DNA damage and chromosome mis-segregation can cause accumulation of cytoplasmic DNA (and RNA) accumulation that triggers type I interferon. This is relevant for auto-immune disorders like Aicardi-Goutieres syndrome or the stimulation of anti-tumor immunity by anti-cancer agents.
The links between neurodegeneration and the response to DNA damage are numerous and ever-growing. As examples, ataxia-telangiectasia is a multi-organ syndrome leading to cerebellar degeneration caused by mutations in ATM, a protein kinase responsive to DNA double-strand breaks and spinocerebellar ataxia with axonal neuropathy-1 (SCAN1) is caused by mutations in TDP1, a DNA tyrosyl-DNA phosphodiesterase whose primary function is to unblock DNA 3’ ends. Furthermore, the metabolism of trinucleotide repeats plays a critical role in the development of neurodegenerative diseases like Huntington’s disease.
We have a wide range of projects that can be broadly grouped in the following three sub-projects:
Featured Paper
Our laboratory discovered a histone modification pathway catalyzed by the RNF8 and RNF168 E3 ubiquitin ligases that culminate in the recruitment of the 53BP1 and BRCA1 DNA repair proteins (Escribano-Diaz et al, 2013; Fradet-Turcotte et al, 2013; Kolas et al, 2007; Stewart et al, 2009; Wilson et al, 2016). Furthermore, we recently discovered an effector of the 53BP1 pathway, named shieldin that likely plays a central role in blocking or reversing end-resection (Noordermeer et al, 2018; Setiaputra & Durocher, 2019). We are deeply invested in understanding how the 53BP1-RIF1-shieldin pathway regulates DNA repair.
The 53BP1 pathway is antagonized by the BRCA1 tumor suppressor and loss of 53BP1 restores homologous recombination and PARP inhibitor resistance in BRCA1-mutated cells and tumors. This striking relationship between BRCA1 and 53BP1 fascinates us (Escribano-Diaz & Durocher, 2013; Noordermeer et al., 2018). We are currently interested in understanding how the BRCA1 E3 ligase activity promotes DNA repair, based on tantalizing observations made recently (Sherker et al, 2021).
Another area of interest is non-homologous end-joining, the DNA double-strand break repair pathway that ligates ends together. We recently identified the SWI/SNF ATP-dependent translocase ERCC6L2 as participating in canonical end-joining (Olivieri & Durocher, 2021) but exactly how ERCC6L2 participates in DNA repair is unknown. As mutations in ERCC6L2 cause bone marrow failure, understanding how ERCC6L2 promotes DNA repair may help understand the role of DNA double-strand breaks in hematopoietic lineage development and may provide therapeutic avenues.
Lastly, we are interested in understanding how endogenous DNA double-strand breaks are formed during the various phases of the cell cycle. We contend that answering this question is essential if we are to harness synthetic lethality approaches with DNA repair modulators.
Ever since the early studies on the effects of radiation on cell division, we know that mitotic cells have a highly specialized DNA damage response. Indeed, the presence of DNA damage in mitosis does not slow down cellular division unless it impacts the formation of functional kinetochores, thus triggering a spindle assembly checkpoint. The highly idiosyncratic DNA damage response in mitosis involves suppression of DSB repair by HR and NHEJ and is also characterized by a blunted RNF8 signaling pathway. We and others contributed to delineating how mitotic kinases suppress the 53BP1 pathway (Orthwein et al, 2015). We are fascinated by this cellular rewiring of the genome maintenance pathways and are excited by the prospect of manipulating the mitotic response to DNA damage for potential therapeutic benefits.
We reported in 2021 the discovery that the CIP2A-TOPBP1 complex acts as a mitotic DNA damage response factor (Adam et al, 2021). Furthermore, loss of CIP2A or the CIP2A-TOPBP1 interaction causes lethality in BRCA1- and BRCA2-deficient cells, identifying this complex as a candidate drug target against HR-deficient cancers. Our data suggests that CIP2A-TOPBP1 may bridge broken ends together to promote the accurate segregation of acentric chromosome fragments.
We are currently focused on understanding CIP2A-TOPBP1 biology: what types of lesions does this complex respond to? How is it integrated with other mitotic processes such as MiDAS, ultrafine bridges resolution, DNA replication termination? What are the pathways that contribute to its recruitment? Are there downstream effectors? What is the biochemistry of the complex? Can this complex be targeted for therapeutic benefit? We are also interested in other aspects of the mitotic DNA damage response and we are excited to explore this biology.
Cancer can be viewed as a disease of the corrupted genome, and we are interested in identifying new genetic vulnerabilities in cancer cells that relate to genome maintenance. We have reported that the APE2 nuclease and CIP2A are essential for the viability of BRCA1/2-deficient cells (Adam et al., 2021; Alvarez-Quilon et al, 2020) and have recently shown, in collaboration with Repare Therapeutics that inhibition of the PKMYT1 kinase, which is a negative regulator of CDK1, is a vulnerability in cell with amplification of CCNE1, the cyclin E1-coding gene (Gallo et al, 2022). We are continuing our work on drug target discovery in oncology and are also working to understand the biology of recently identified drug targets.
Another emerging area of interest is to assess or implement new therapeutic modalities that would enable the modulation of DNA repair for therapeutic purposes. One approach that we are currently evaluating is best described as induced proximity and involves the generation of new protein-protein interactions. Indeed, as biological processes often depend on proteins interacting together, it seems intuitive that generating new protein-protein interactions may allow one to modulate biological process, possibly in a manner that could be harnessed therapeutically.